
Your doctor can order a genetic test for FSHD. Before seeking a test, consult a genetic counselor to make sure you fully understand the process and have considered how you and your family will respond to the information revealed by the test results.
- To understand some of the issues that may come up, read our article FAQs about genetic testing for FSHD, by genetic counselor and FSHD expert Julie Cohen, MSc.
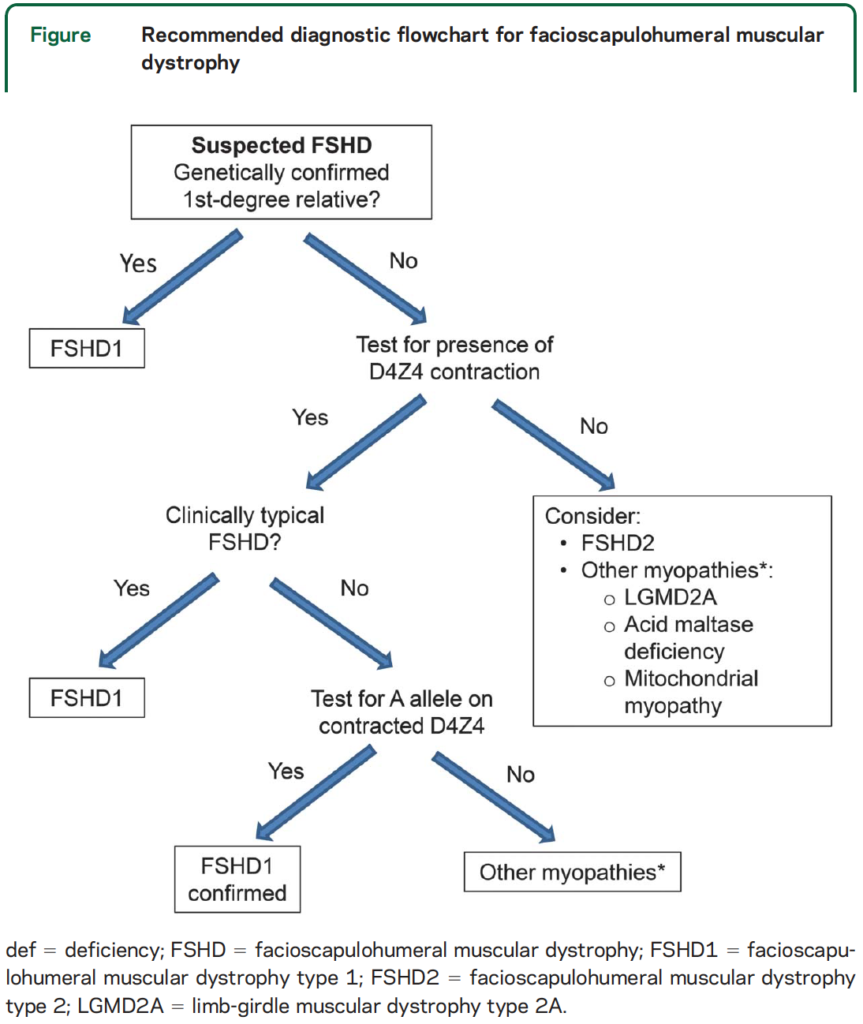
- The genetic cause of FSHD is complicated! Learn more here.
Commercial genetic tests are available for FSHD Type 1 and Type 2. If you already have a family member who has been tested, find out what type of FSHD they had (get a copy of their report if possible), as you will only need to be tested for that type. If you don’t have a family member who has been diagnosed, you will usually be tested for Type 1 first because around 95 percent of cases are Type 1. If you test negative for Type 1, you may inquire about obtaining a test for FSHD Type 2 or for a broader panel of neuromuscular conditions.
- To understand the types of FSHD genetic testing that are available, and the difference between research and clinical testing, read Genetic testing for FSHD-A new frontier.
Insurance companies may refuse to cover genetic testing in family members who may be asymptomatic but at risk of having, and passing along, the FSHD gene. If that happens, use this letter template to file an appeal:
If a clinical diagnostic test is not for you, you can learn about your genetic status by participating in a research study such as the U.S. National Registry or the laboratory of Peter Jones. Visit our Clinical Trials page for information on enrolling in these studies.
Lowering barriers to genetic testing for FSHD
Currently, one of the eligibility criteria for volunteering in an FSHD clinical trial is positive confirmation of the disease by a clinically approved genetic test. Unfortunately, many patients and families who are trying to get tested have faced barriers, from doctors who don’t know how to order the test to insurance companies that balk at paying for it.
In response to this challenge, the FSHD Society, in collaboration with industry sponsors, has established a clinically approved genetic testing program for the FSHD community in the US.
DISCLAIMER: Information provided by the FSHD Society does not imply an endorsement of any of the drugs, procedures, treatments, or products discussed. Please consult your own healthcare provider about any medical interventions.